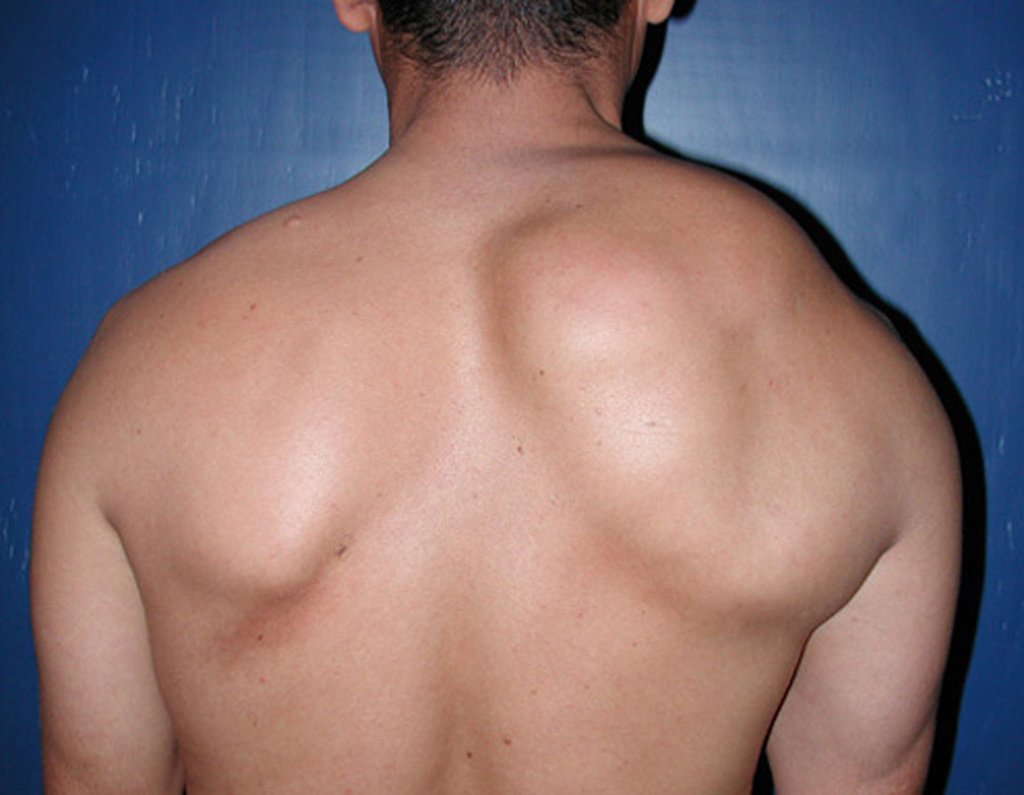
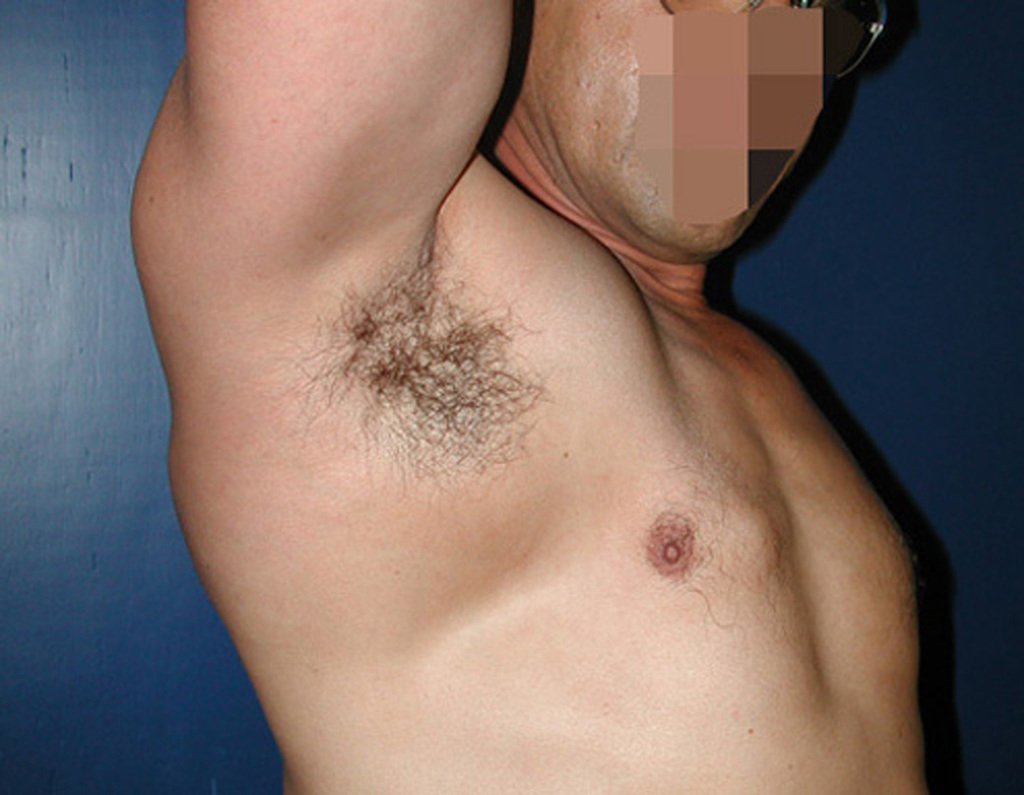
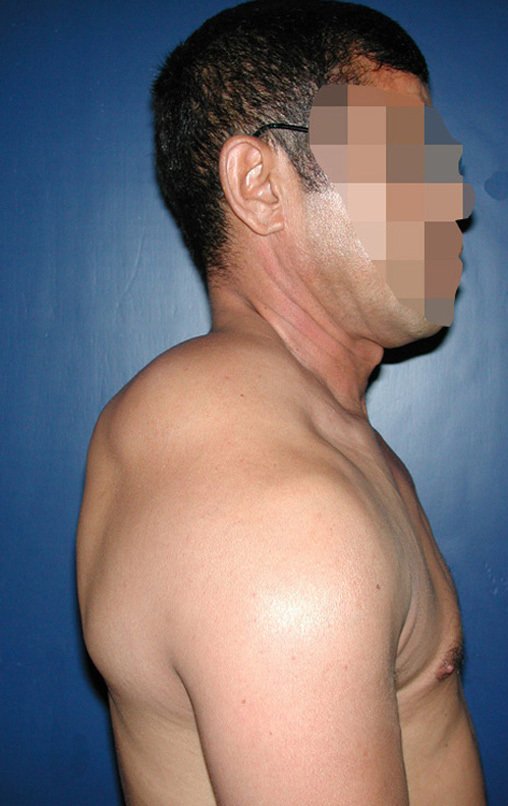
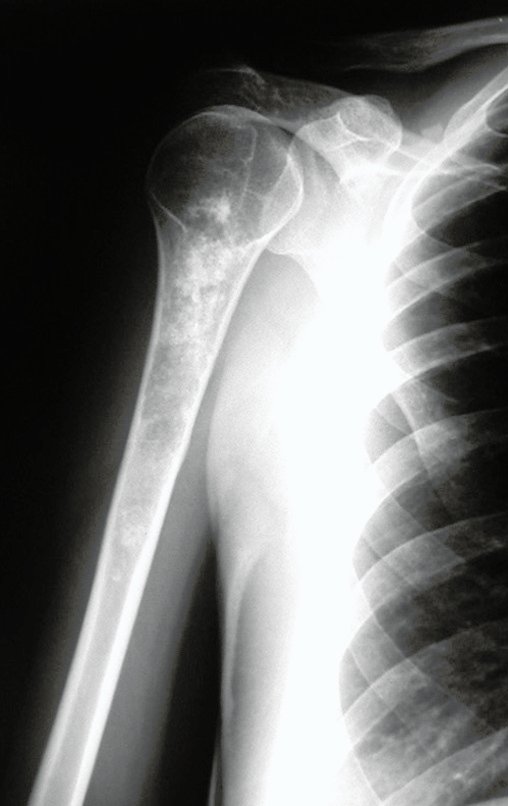
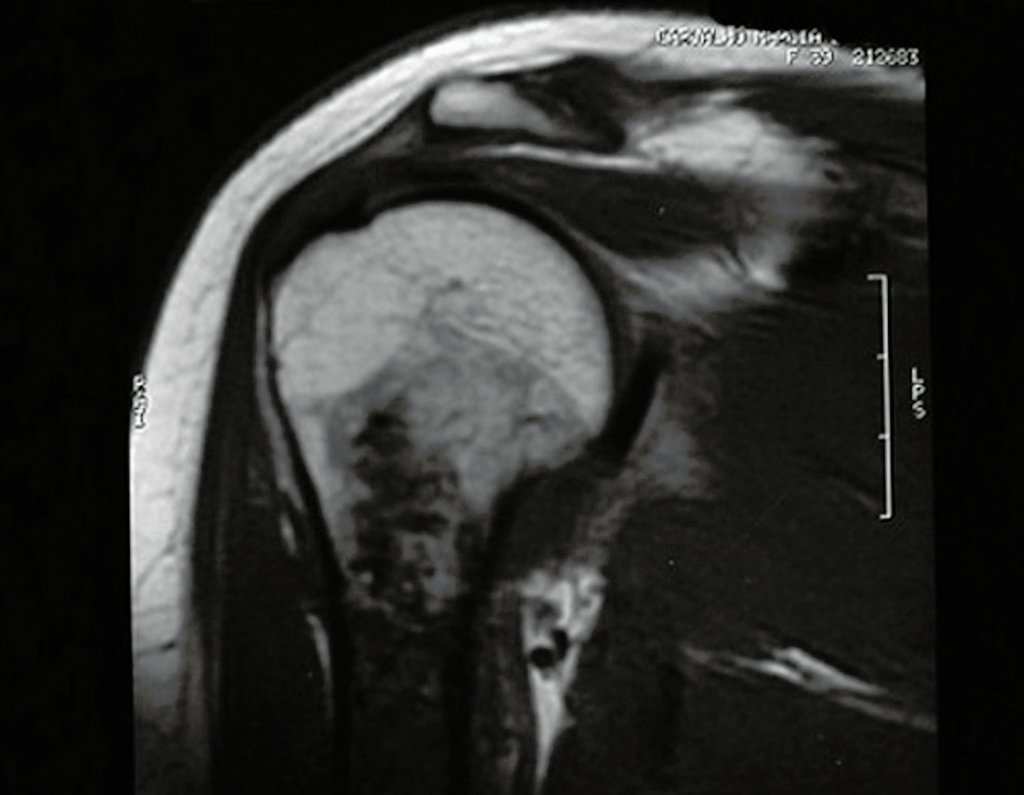
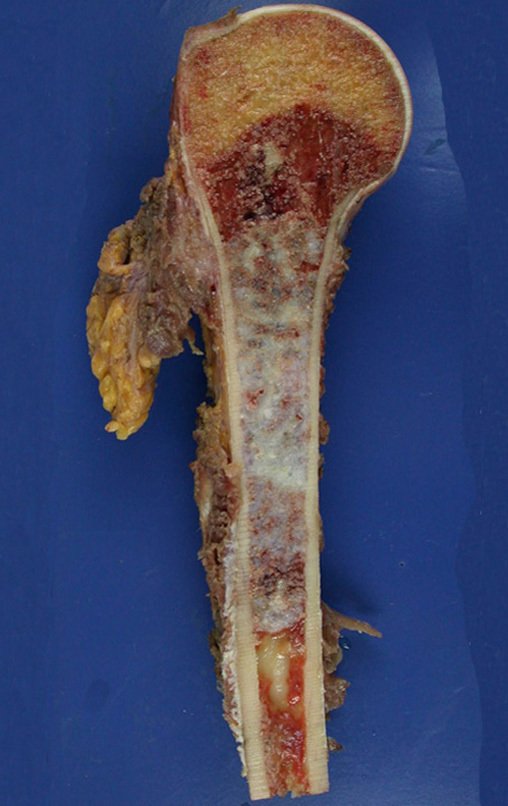
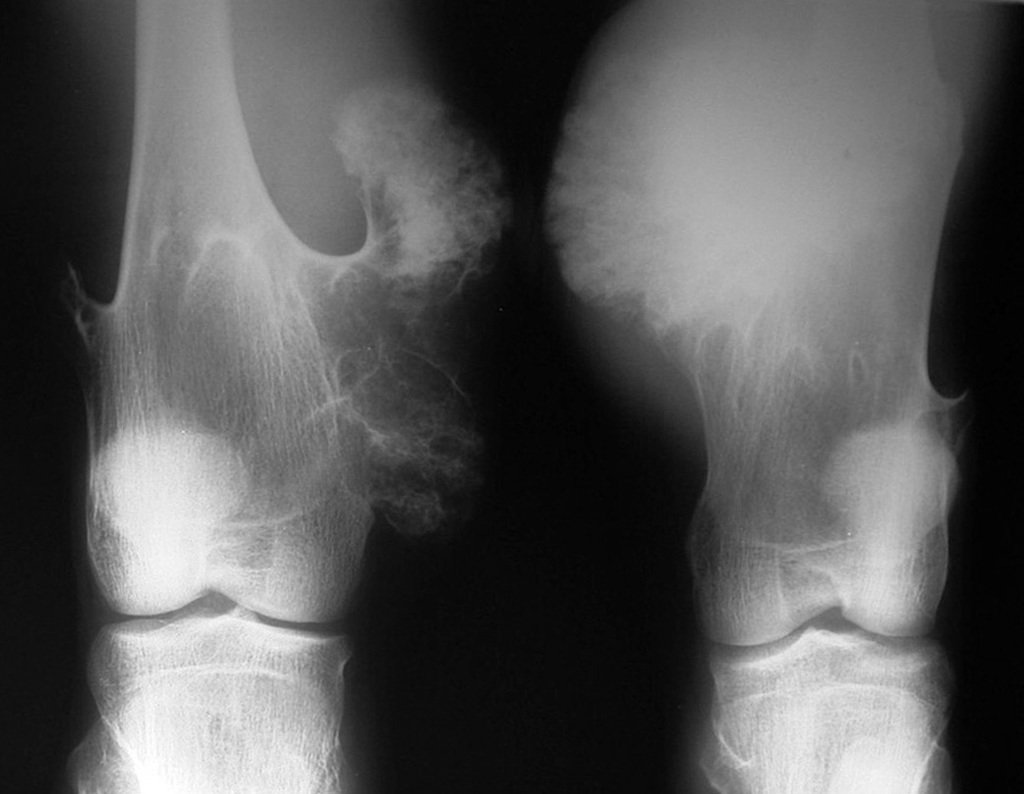
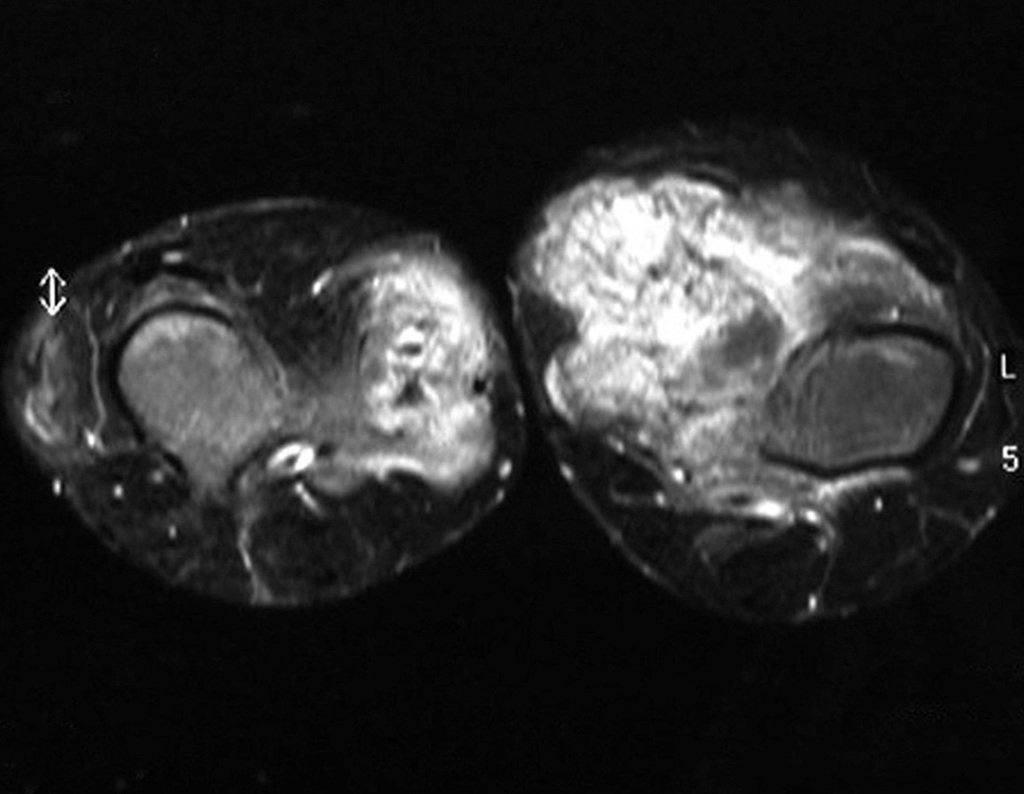
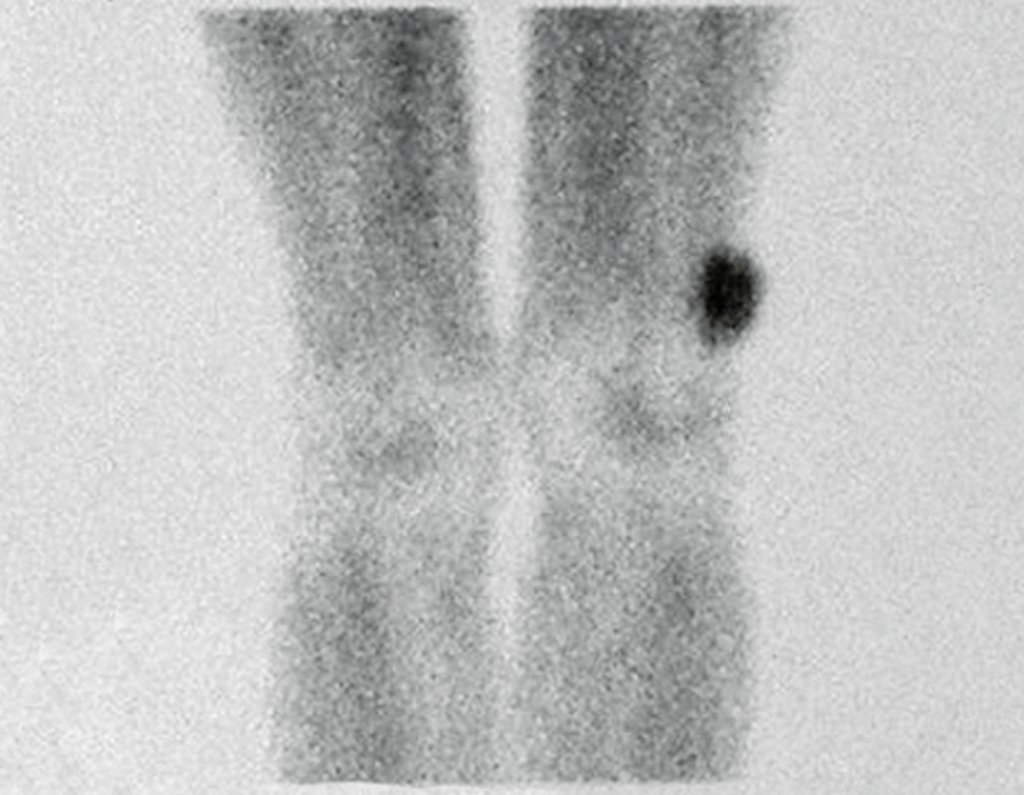
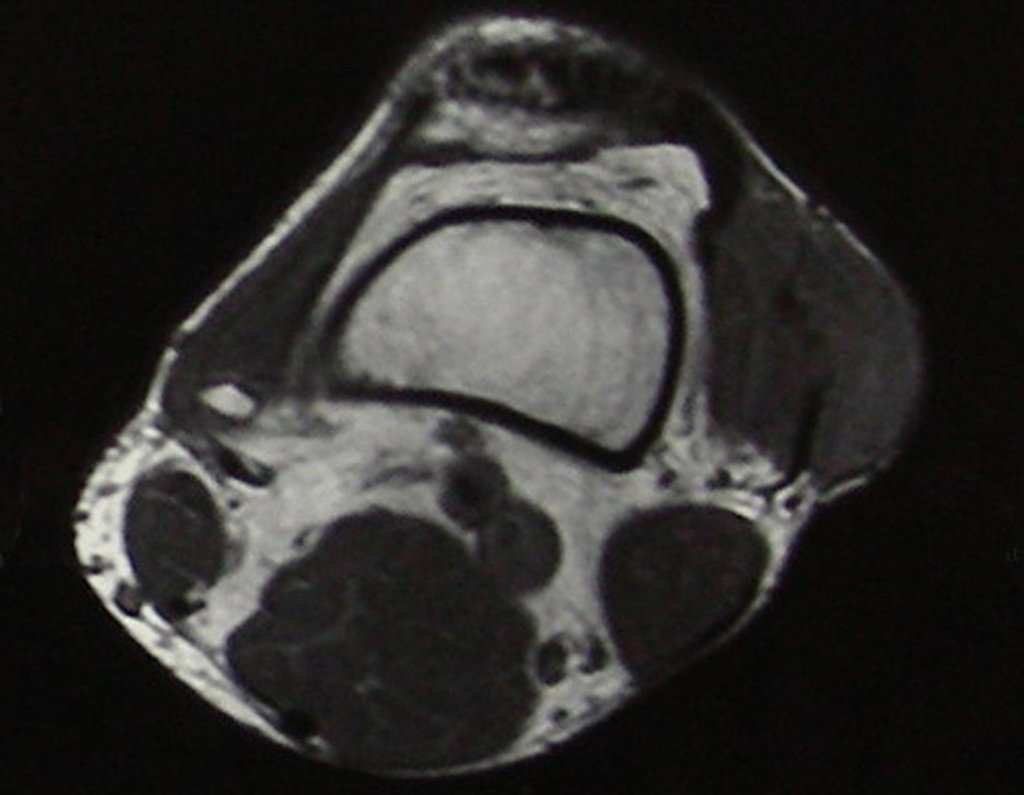
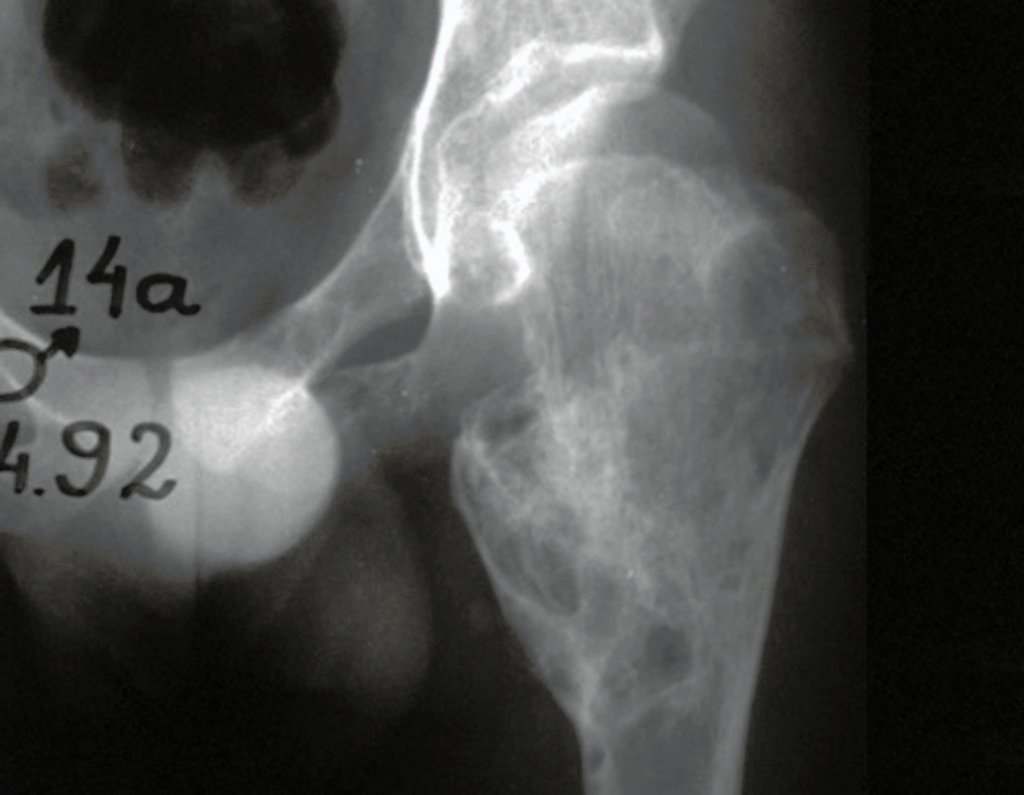
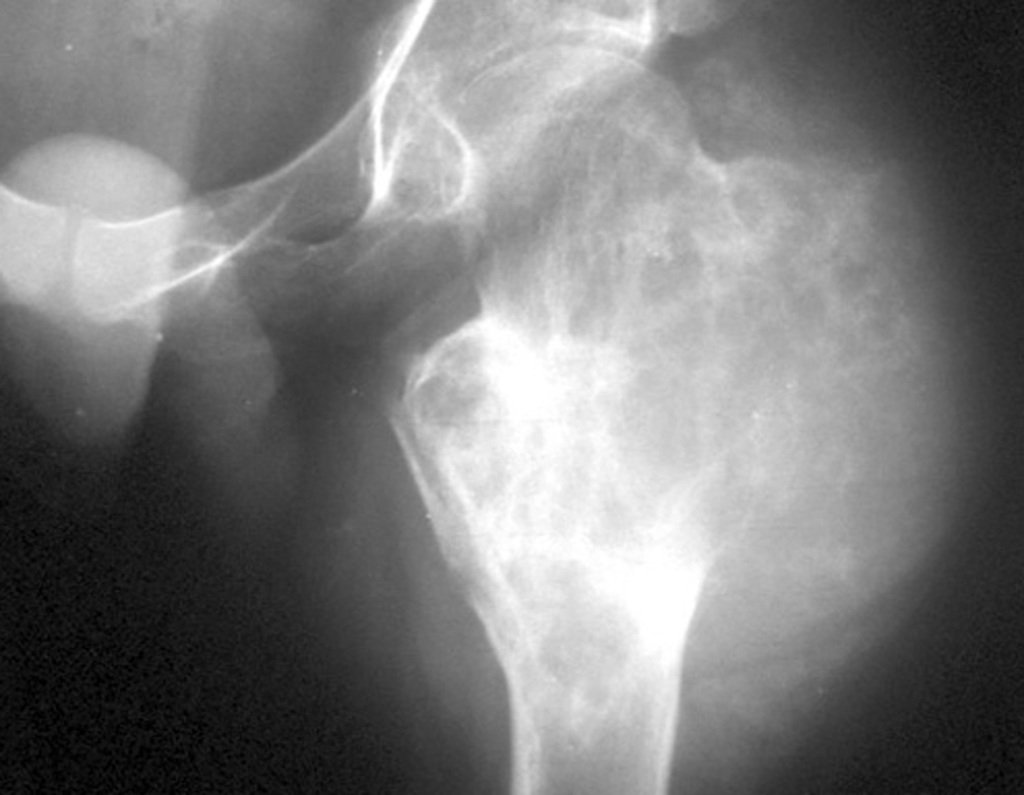
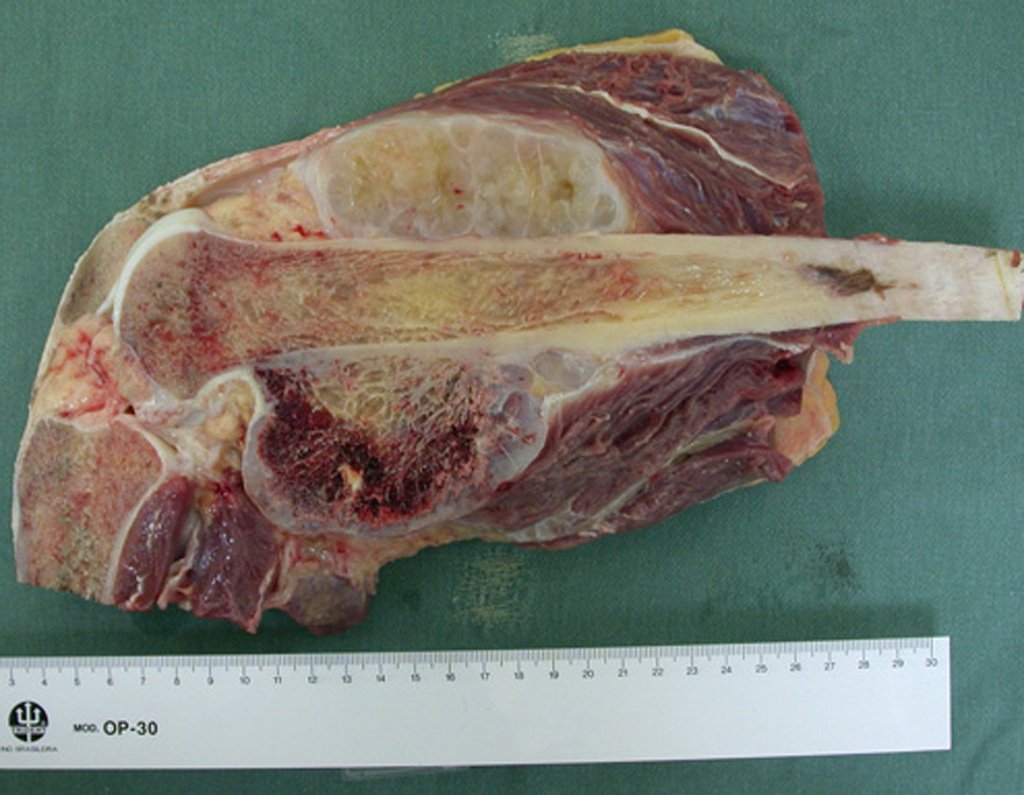
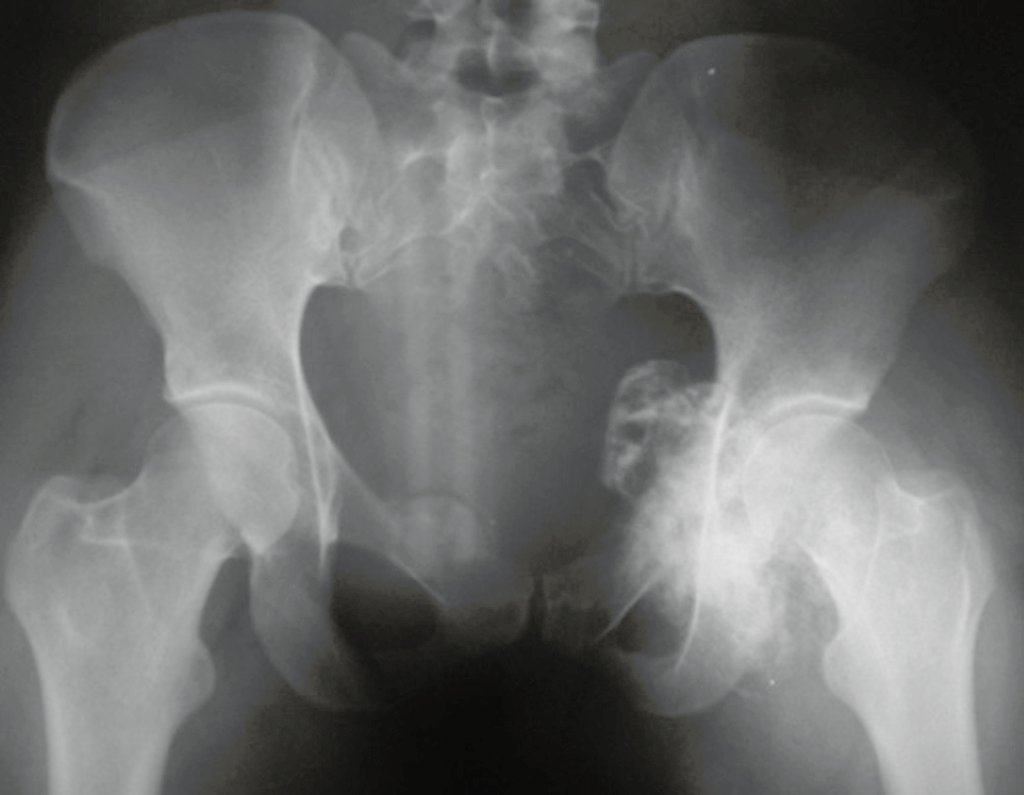
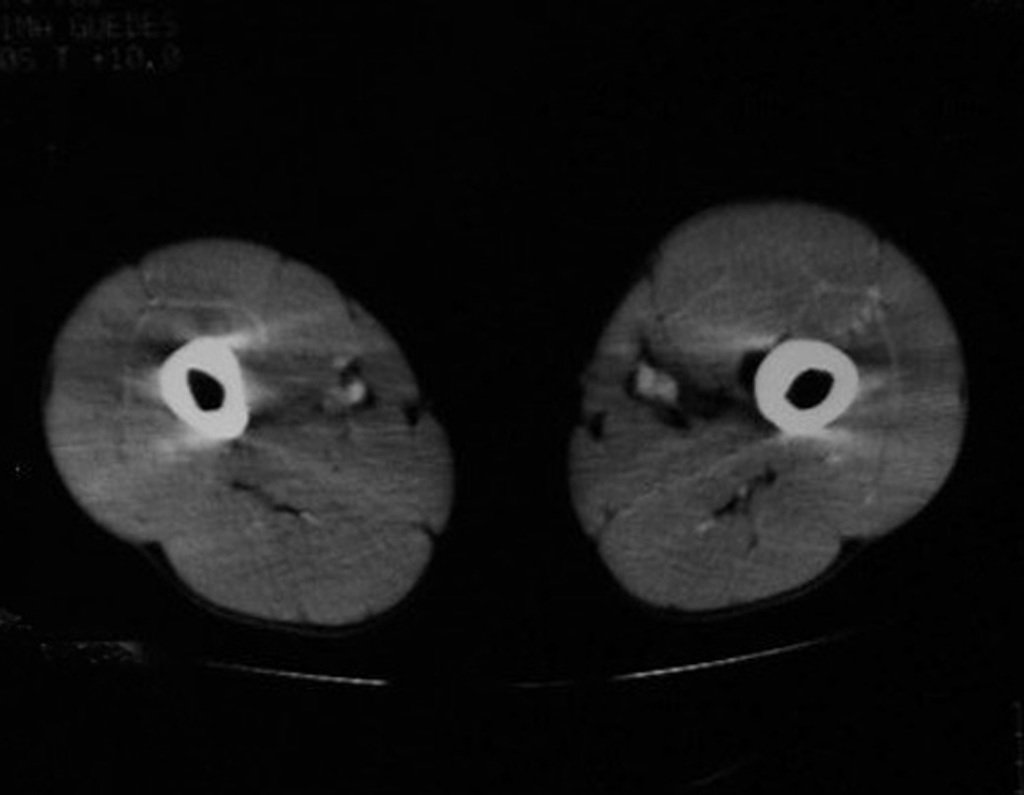
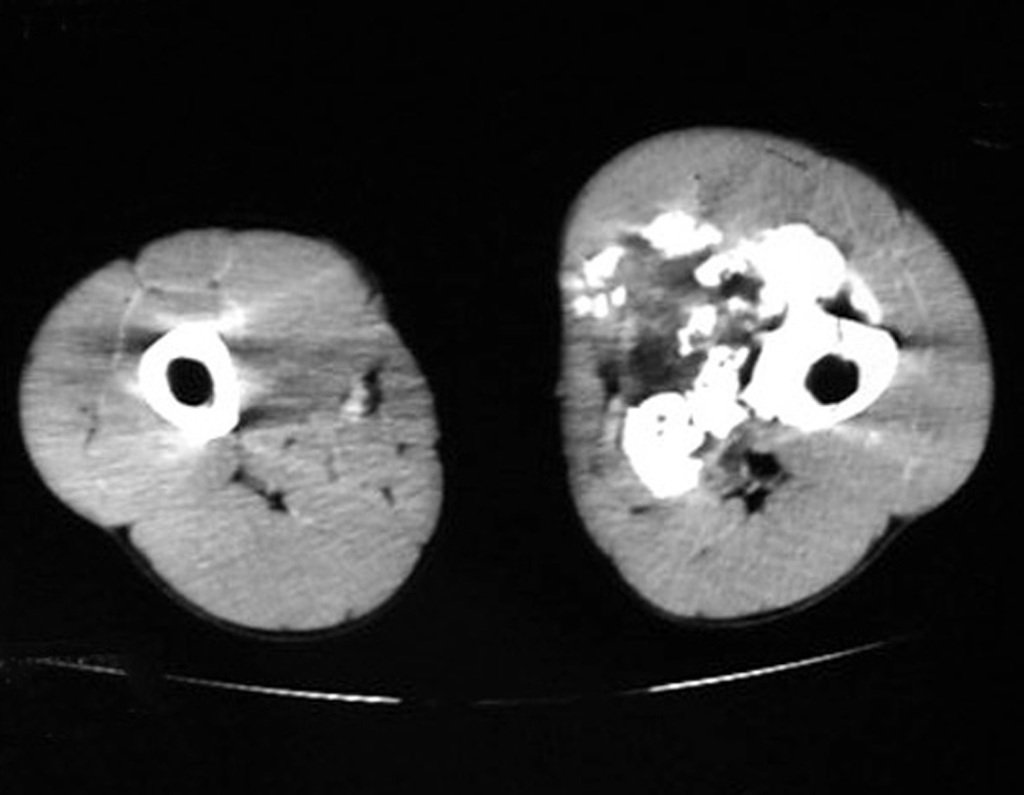
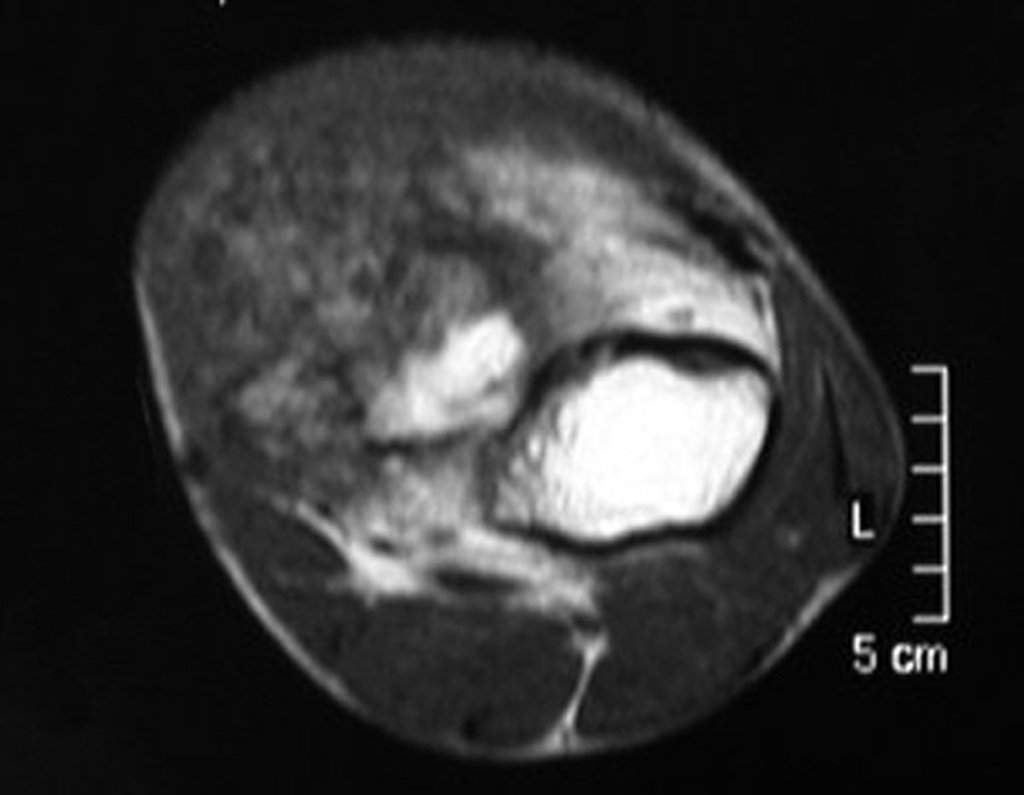
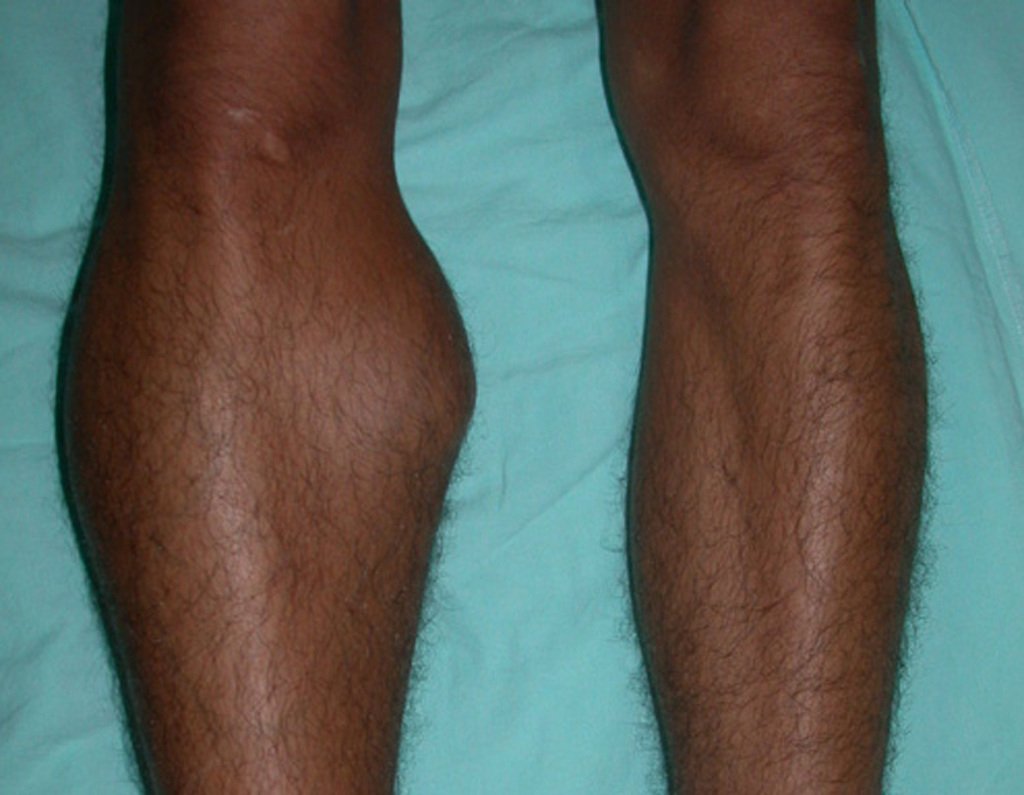
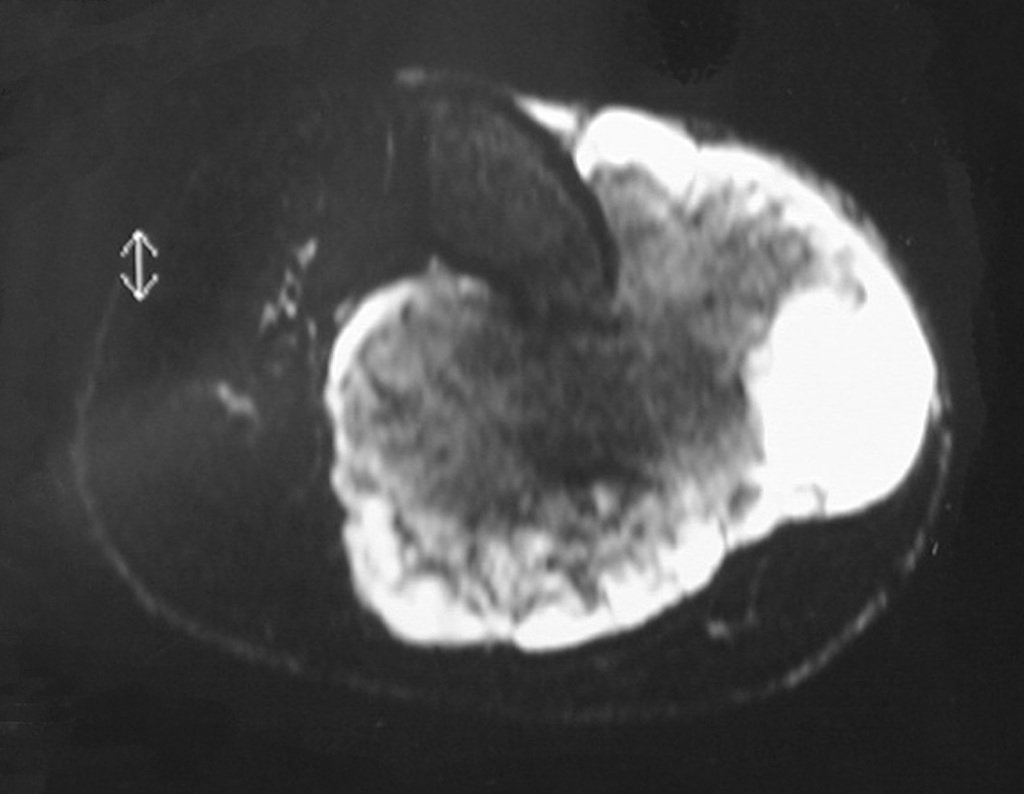
Diagnóstico Diferencial:
Apresenta diagnóstico diferencial com a miosite ossificante, o fibroma condromixóide, o T.G.C., o linfoma não Hodgkin6,23,29 e com o cisto ósseo aneurismático, por seu caráter multiloculado. Histológicamente, o subtipo justacortical assemelha-se ao condroma, ao osteocondroma, ao condroblastoma e ao osteossarcoma de superfície16.
O condrossarcoma de células claras tem condrócitos malignos com citoplasma claro, células gigantes tipo osteoclasto e formação de osso reativo intralesional causando confusão com osteossarcoma.
O condrossarcoma mesenquimal é formado por ilhas de cartilagem hialina bem diferenciada circundada por lâminas de células pequenas e redondas, que lembram hemangiopericitoma e sarcoma de Ewing 14.
O condroma central dos ossos longos, o condrossarcoma e o infarto ósseo são muitas vezes de difícil diferenciação, necessitando acompanhamento clínico e radiográfico para avaliar a progressão ou não da lesão, antes de definir a conduta. A biópsia muitas
vezes não é definitiva para o diagnóstico12,23,29.
Tratamento:
O tratamento do condrossarcoma é cirúrgico25, devendo-se eleger uma ressecção ampla, incluindo o trajeto da biópsia13,21.
A radioterapia é ineficaz6 no controle desta neoplasia. Para as lesões de alto grau pode-se discutir indicação de quimioterapia utilizando o protocolo para sarcomas de grandes células, baseada em antraciclicos9999. Para o condrossarcoma mesenquimal, que apresenta predomínio de células pequenas e indiferenciadas, a quimioterapia quando indicada recai sobre o protocolo de tratamento do Tumor de Ewing.888
Em ambos os casos a resposta à quimioterapia costuma ser ruim6. O tratamento desta neoplasia deve ser particularizado para cada subtipo clínico:
– Condrossarcoma central apresenta altos índices de cura com a cirurgia adequada, portanto não se pode subestimar o seu tratamento com curetagem intralesional,mesmo seguida de métodos adjuvantes complementares, seja com fenol, nitrogênio líquido, eletrotermia ou laser CO2 21.
Desta maneira, nos casos de dúvida diagnóstica entre condroma e condrossarcoma grau I é preferível observar a evolução desta lesão, pois é sabido que a biópsia não será conclusiva, já que o diagnóstico diferencial histológico entre condroma e condrossarcoma grau I é difícil.
Em alguns casos, estas lesões podem ser tratadas com cirurgias conservadoras sem a realização de biópsia prévia21.
Quando os exames de imagem: radiografia, tomografia e ressonância magnética, mostram uma lesão central, sem erosão da cortical interna, de achado casual e indolor deve-se reavaliar inicialmente dentro de três meses, estando inalterado repete-se no período de seis meses e se a lesão permanecer inalterada, programa-se reavaliações anuais.
Se, em qualquer momento, houver alteração do quadro clínico ou de imagem, deve-se tratar como condrossarcoma central, realizando-se a ressecção ampla da lesão e reconstrução com endoprótese não convencional, osteossíntese com enxerto autólogo ou homólogo ou cirurgia ablativa conforme a necessidade de cada caso.
Na experiência destes autores é desnecessário operar um condroma indolor, quando é achado casual, sem caracteres de agressividade radiográfica. Realizar uma curetagem intralesional, com adjuvante local e enxerto ou cimento, não dispensará a necessidade de observação cuidadosa. Caso o exame anatomopatológico de toda a curetagem revele que se tratava de condrossarcoma, será muito pior re-operar esta região já manipulada cirurgicamente.
Há vários casos de “condroma” em que a histologia da curetagem intralesional corroborou o aspecto da biópsia de “condroma” e no entanto tiveram evolução desfavorável. No acompanhamento destes pacientes os exames de imagem revelaram que estava havendo “nova” lesão no local e que se tratava agora de condrossarcoma.
Nestas curetagens pode ocorrer disseminação local, à distância e até desdiferenciação do condrossarcoma, piorando significativamente o prognóstico.
– Condrossarcoma justacortical, o tratamento é essencialmente cirúrgico, podendo-se realizar a ressecção parcial parietal EXEMPLO quando possível, procedimento eficaz e de menor morbidade em relação a ressecção segmentar.
– Condrossarcoma periférico, secundário à osteocondroma, deve-se tomar cuidado principalmente com a superfície da lesão, que apresenta anaplasia.
O perimísio dos tecidos moles ao redor deve ser removido como margem oncológica, para evitar a recorrência local.
É importante ressaltar que quando ocorre crescimento de uma exostose óssea após a maturidade esquelética, calcificação heterogênea, capa cartilaginosa espessa, sem relação com atrito ou trauma, provavelmente trata-se de um condrossarcoma.
Nesta situação, uma amostra de biópsia negativa não exclui a possibilidade de malignidade no restante da lesão, devendo-se realizar a cirurgia de ressecção com margem oncológica, com especial atenção à superfície da lesão.
– Condrossarcoma Mesenquimal, além da necessidade do controle local com a cirurgia ampla, podem eventualmente ter indicação de tratamento quimioterápico complementar9999.
– Condrossarcoma Desdiferenciado, como o Condrossarcoma de Células Claras, deve-se realizar o controle local com a cirurgia ampla e quimioterapia com cisplatina e doxorubicina9999.
Complicações:
A curetagerem intralesional de condrossarcoma pode levar a recorrência local da doença e a desdiferenciação histológica mais agressiva.
Nos casos de condrossarcomas desdiferenciados, as metástases hematogênicas para os pulmões são freqüentes, podendo apresentar disseminação linfática e recidiva local29. Muitos condrossarcomas apresentam tendência de disseminação local14, atingindo tamanhos enormes e tornando-se inoperáveis, causando a morte por compressão ou complicações desta propagação local.
A recidiva local aumenta a incidência de metástases pulmonares21.
Bibliografia
1. ACKERMAN, L.V.; SPJUT, H.J. Tumors of bone and cartilage. Atlas of tumor pathology. Washington,Air Force Inst. Pathology, 1962, fasc, 4.
2. CANALE, S.T. Cirurgia ortopédica de Campbell.Barueri: Manole; 2006
3. DAHLIN, D.C. Tumores óseos . Barcelona: Ediciones Toray S/A; 1982
4. DORFMAN, H.D.; CZERNIAK, B. Bone tumors. St Louis, C.V. Mosby Co., 1997, cap. 7, p.410.
5. EDEIKEN, J.; HODES, P.J. Diagnóstico radiológico de las enfermedades de los huesos. Buenos Aires, Panamericana, 1977, cap. 15.
6. ETCHEBEHERE, M. Tumores cartilaginosos malignos: Condrossarcomas. In: Camargo O.P. Clínica Ortopédica. Rio de Janeiro: Med si; 2002. p. 753-759
7. FELDMAN, F. Cartilaginous tumors and cartilage-forming tumor like conditions of the bonés and soft tissues. In: Diseases of the Skeleton System (Roentgen Diagnosis). Part. 6 – Bone Tumors, New York, Springer-Verlag, 1977,p.177.
8. FLETCHER, C.D.M., Unni K.K., OMS – Merters F. (Eds.): World Health Organization. Classification of Tumors. Pathology and Genetics of Tumors of Soft Tissue and Bone. IARC Press: Lyon 2002.
9. GREENSPAN, A. Radiologia ortopédica. Rio de Janeiro: Guanabara; 2001.
10. HENDERSON, E.D.; Le PAGE, G. A. Apud FELDAMAN, F. Cartilaginius tumors and cartilage forming tumor like conditions of the bone and soft tissues. In: Disease of the Skeletal System (Roentgen Diagnosis).
Part. 6 – Bone tumors, New York, Springer Verlag, 1977, p.182.
11. HUVOS, A.G. Bone tumors Diagnosis, Treatment and Prognosis. Philadelphia, W. B. Saunders Co., 1979, p. 13.
12. JAFFE, H.L. Tumores y estados tumorales oseos y articulares. México: La Prensa Medica Mexicana;1966.
13. JESUS-GARCIA, R. – Reynaldo Jesus-Garcia
14. LICHTENSTEIN, L. Barcelona: Talleres Gráficos Ibero-Americanos; 1975.
15. LICHTESTEIN, L. Bone Tumor. 4 Ed St. Louis,C.V. Mosby Co., 1972, cap. 15.
16. LICHTESTEIN, L.; BERNSTEIN, D. Unusual benign and malignant chondroid tumors of bone. Cancer, 12:1142, 1959.
17. MARCOVE, R.C. Condrosarcoma: Diagnóstico y tratamiento. In: Clínicas Ortopécias de Norteamérica. Tumores del aparato musculosquelético. Buenos Aires, Panamericana, 1977, cap. 7.
18. MARCOVE, R.C. et al. Chondrosarcoma of the pélvis and upper end of the femur. Na analisys of factors influencing survival time in113 cases. J. Bone Joint Surg., 54A:61, 1972.
19. MARCOVE, R.C.; SHOJI, H,; HARLEN, M. Altered carbohidrate metabolism in cartilaginous tumors. Contemp. Surg. 5:53, 1974.
20. McFARLAND, G.B.Jr.; McKINLEY, L.M.; REED, R.J. Dedifferentiation of low grade chondrosarcomas. Clin. Orthop., 122:157, 1971.
21. MENENDEZ, L.R. Orthopaedic knowledge update: Actualizaciones en cirugía ortopédica y traumatología. Barcelona: Ars Medica; 2003.
22. O’NEAL, L.W.; ACKERMAN, L. V. Chondrossarcoma of boné. Cancer, 5:551, 1952.
23. PRÓSPERO, J.D. Tumores Ósseos. São Paulo, Roca, 2001, cap. II.
24. ROBBINS. Patologia estrutural e funcional. Rio de Janeiro: Guanabara; 1996.
25. ROMSDAHL, M.; EVANS, H.L.; AYALA, A.G. Surgical treatment of chondrosarcoma. In: Managment of primary bone and soft tissue tumors. Chicago, Year book med. Publisher Inc., 1977, p. 125.
26. ROMSDAHL, M.; Evans, H.L.; Ayala, A.G. Surgical treatment of chondrosarcoma. In: Managment of primary bone and soft tissue tumors. Chicago. Year book med. Publisher Inc., 1977, p.125.
27. SALVADOR, A.H.; BEABOUT, J.W.; DAHLIN, D.C. Mesenchimal chondrosarcoma. Cancer, 28:605, 1971.
28. SCHAJOWICZ, F. Justacortical Chondrosarcoma. J. Bone Joint. Surg., 59B:473, 1977.
29. SCHAJOWICZ, F. Tumores y Lesiones Seudotumorales de Huesos y Articulaciones. Buenos Aires: Editora Médica Panamericana; 1982.
30. TORNBERG, D.N.; RICE, R.W.; JOHNSTON, A.D. The ultrastructure of chondromyxoid fibroma.Clin. Orthop. Rel. Research, 95:295, 1973.
999. J Clin Oncol 30:abstrat 100:23,2012(maluf)
888. Buzaide, A.C.; Maluf, F.C.; Rocha Lima, C.M.
Manual de Oncologia Clinica do Brasil. Dendrix Edição e Design ltda. São Paulo (XI) Sarcomas Ósseos do Adulto, 560-79. 2013